Was ist MAP (MUTYH-assoziierte Polyposis)?
Im Jahre 2002 wurde eine neue Form der adenomatösen Polyposis entdeckt, die als MAP (MUTYH-assoziierte Polyposis) bezeichnet wird.
Klinisch ist die MAP im Wesentlichen vergleichbar mit der durch APC-Mutationen verursachten attenuierten FAP (siehe AFAP).
Im Unterschied zu der bisher bekannten autosomal-dominant erblichen und durch Mutationen im APC-Gen ( auf Chromosom 5) verursachten FAP und AFAP wird die MAP durch Veränderungen im MUTYH-Gen (auf Chromosom 1) verursacht und folgt einem autosomal-rezessiven Erbgang (siehe Abbildung).
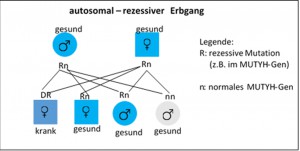
Dieses bedeutet, dass die Polyposis-Erkrankung erst auftritt, wenn beide Kopien des MUTYH-Gens (die vom Vater und die von der Mutter geerbte Kopie) eine Veränderung aufweisen.
Kennzeichnend für den autosomal-rezessiven Erbgang ist, dass beide Eltern gesund sind und oft ein Einzelpatient oder mehrere Geschwister in der Familie erkrankt sind.
Eine MAP wird bei etwa 15-20% der Patienten mit einer milden Verlaufsform der adenomatösen Polyposis diagnostiziert.
Wegen des hohen Krebsrisikos wird MAP-Patienten (also Trägern von Mutationen in beiden Kopien des MUTYH-Gens) das gleiche Früherkennungsprogramm wie Kindern von Patienten mit der APC-assoziierten AFAP empfohlen. Dieses umfasst eine jährliche komplette Koloskopie ab dem Alter von 18 Jahren. Bei Feststellung von Polypen erfolgt das weitere Vorgehen in Abhängigkeit vom klinischen Befund.
Abbildung: Mareike Demand
Autor: Dr. Waltraut Friedl, Institut für Humangenetik der Universität Bonn
Letzte Überarbeitung: November 2014